Homocystinuria is associated with the following symptoms and side effects if not treated :
- Downward dislocation of lens (ectopia lentis)
- Mental retardation
- Strokes
- Severe nearsightedness
- Convulsions
- Mental problems
- Bone problems ( osteoporosis, scolliosis)
- Formation of blood clot that is known as Thromboses.
- Kidney problem
- coronary artery disease
Clinical abnormalities in CbS deficiency (Mudd et al. [9])
Symptoms
- A family history of homocystinuria
- Nearsightedness
- Flush across the cheeks
- Tall, thin build
- Long limbs
- High-arched feet (pes cavus)
- Knock-knees (genu valgum)
- Pectus excavatum
- Pectus carinatum
- Mental retardation
- Psychiatric disease
Ocular
| Frequent | Less Frequent |
| Ectopia lentis Iridodenesis Myopia |
Glaucoma Optic atrophy Retinal degeneration Retinal detachment Cataracts Corneal abnormalities |
Skeletal
| Frequent | Less Frequent |
| Osteoporosis Biconcave(“codfish”) vertebrae Scoliosis Increased length of long bones Irregular widened metaphysis Metaphyseal spicules Abnormal size and shape of epiphysis Growth arrest lines Pes cavus High-arched palate |
Arachnodactyly Enlarged carpal bones Abnormal bone age Pectus carinatum or excavatum Genu valgum Kyphosis Short fourth metacarpal |
Central nervous system
| Frequent | Less frequent |
| Mental retardation Psychiatric disturbances Extrapyramidal signs |
Seizures Abnormal electroencephalogram |
Vascular system
| Frequent |
| Vascular occlusions Malar flush Livedo reticularis |
Other involvement
| Fair, brittle hair Thin skin Fatty changes in liver Inguinal hernia Myopathy Endocrine abnormalities Reduced clotting factors |
It is worth emphasizing that the patient with CbS deficiency is normal at birth and if left untreated, progressively develops the full-blown clinical picture [29].
Ophthalmological manifestations
Ectopia lentis is the hallmark and most consistent clinical finding in classical homocystinuria, indeed many cases have been diagnosed because of it [20,30 ]. It occurs in 85% of CbS deficiency cases [20,28,31]. Ectopia lentis and high myopia ( < 5 Dioptres) are undeniably the major ocular manifestation of classical homocystinuria [32]. The natural history of developing ectopia lentis in classical homocystinuria is such that 70% had ectopia lentis by age 10 years [9].
Disruption of the thickened zonulae fibres causes the lens to dislocate inferiorly or nasally, or both, and is usually bilateral [32-34]. This disruption of the thickened zonulae fibres leads to increased curvature of the lens and thereby to lenticular myopia and astigmatism. Iridodenesis is the quivering of the iris by moving the eye ball and is a sign of the loosening of the lens. Acute pupillary block glaucoma may result from anterior dislocation of the lens [9].
In a study of 34 patients with CbS deficiency, Cruysberg et al reported on the prevalence of ocular manifestation in these patients and found that the highest prevalence were found for myopia > 1D (85%), high myopia > 5D (76%), ectopia lentis (76%), iridodenesis (56%), spherophakia (50%) and very high myopia >10D (50%). Ectopia lentis, associated with late diagnosis of CbS deficiency, was identified as the major risk factor for other ocular complications, including strabismus (24%), dense cataract (21%), acute pupillary block glaucoma (19%), retinal detachment (15%), and unilateral blindness (18%) [32].
Cruysberg et al in 1996 also pointed out that there should be a high level of suspicion that high myopia > 5D may be caused by ectopia lentis [35]. In such high myopia, one or the other abnormality of ocular refraction of the axial length, the corneal curvature or the lens, the most likely, must be present. Other clues for lenticular myopia are abnormally rapid progression of myopia, high myopia in children as myopia > 5D is extremely rare in children under 2 years [36], progressive myopia in adult life as high myopia probably affects under 2% of adults [36], and high non-corneal astigmatism with subnormal visual activity [35].
As pointed out by Mudd et al, the data on age of lens dislocation is more representative of the age/date of detection rather than it’s actual occurrence [20]. In homocystinuria, the age of development of high myopia >5D prior to lens dislocation is a better indicator of the start of lens dislocation. On one hand, a normal ophthalmological examination at any age should not be a reason for rejecting the diagnosis of classical homocystinuria [9], and on the other, a high level of suspicion for the diagnosis must be present in those presenting with unusual myopia particularly if combined with skeletal, vascular or central nervous system manifestation [35].
Skeletal abnormalities
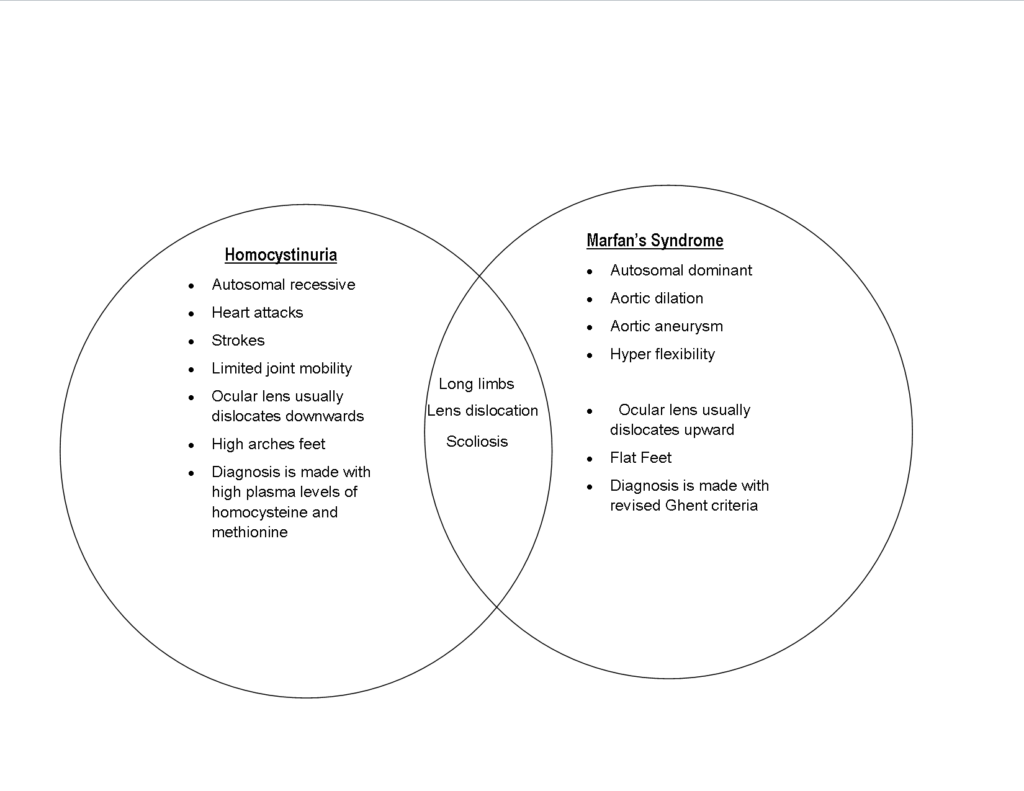
Among the most striking changes in persons with CbS deficiency are those of the skeleton, however, these changes are not evident at birth [37], and usually not in infants and very young children. The appearance of genu valgum and pes cavus [38] are usually the first signs. As puberty nears, dolichostenomelia becomes evident and their limbs appear to grow out of proportion to the trunk, anterior chest wall deformities may occur taking the form of pectus excavatum or carinatum and kyphosis or scoliosis may be present [30,38] The facial appearance may be altered by prominence and protrusion of the upper teeth due to overcrowding and the palate is almost always high arched [38,39]. These skeletal manifestation gives older patients an appearance similar to that of Marfan’s syndrome [28,37,38,40-43]. One of the distinguishing features of classical homocystinuria is the presence of osteoporosis [30].
Osteoporosis, especially of the spine, is the most consistent skeletal abnormality on radiograph. Although it is not present at birth, it has been described as early as at the age of one year [41,44]. The frequency of osteoporosis in CbS deficient cases has been reported to be as high as 90-100% [38,40-42,44], with one exception in which study it was only 35% [44]. Time-to-event curve for radiological evidence of spinal osteoporosis in untreated patients predicts a that 50% have radiological evidence of osteoporosis by age 15 years [9]. Spinal osteoporosis generally occurs earlier in pyridoxine non-responsive cases than in those responsive [20]. Osteoporosis may lead to scoliosis, however, it has also been reported in the absence of osteoporosis [45]. Vertebral collapse may also be from the effects of osteoporosis [14].
Radiologically, there is posteriorly placed biconcavity of the vertebrae (“codfish”) unlike the biconcavity typical of osteoporosis as pointed out by Brenton et al. [38]. The hands and feet are also the site of several radiological changes with prominent growth arrest lines are often present in the distal tibia [45] and curiously, development of the lunate has been selectively retarded in some cases [41,42,46]. The bone age is not usually altered, although on occasions, it may be either advanced or delayed [36].
Vascular manifestations
The cardinal vascular sign in classical homocystinuria is thromboembolism, affecting both large and small arteries and veins [30]. It is also the most striking cause of serious complications and mortality in CbS deficiency [9,20]. Vascular occlusion may occur at any age and the natural history of clinically detected thromboembolic events is such that 30% would have had an event by age 20 years and this further increases to 50% by age 30 years [20]. Beyond the age 10 years there was one event per 25 years and the event rate was constant.
Thrombophlebitis is the most frequent complication often leading to fatal pulmonary emboli [14,40,44,47-49] or chronic cor pulmonale [50,51]. Thrombosis of the great veins: inferior vena cava, femoral veins, iliac veins, renal veins [46] and vena portae [15], have all been reported in the literature.
Serious complications of thromboembolism have included optic atrophy secondary to occlusion of optic artery, hemiparesis, severe hypertension due to renal infarcts, seizures or focal neurological signs due to cerebral thrombi. Sagittal sinus thrombosis was the presenting feature in an otherwise asymptomatic adolescent [52]. Mudd et al in their international survey of 629 cases of CbS deficiency reported that only 4% of all thromboembolic events produced myocardial infarction [20].
It is to be noted here that during their young adult years, the chance that an untreated CbS deficient individual previously free of thromboembolic events would have such a clinically detected event during the subsequent year was 1:25 [9]. However, with the advent of ultrasonography, many patients have been shown to have signs of early vascular disease even in the absence of ischaemic symptoms. [9,53]. Mudd et al also surveyed 586 major surgeries and eye operations in CbS deficient cases to establish postoperative thromboembolic complications and found six fatal cases out of 25 postoperative thromboembolic events that developed [20]. Hence, if absolutely indicated, the great majority of surgery may be conducted without vascular complications in patients with classical homocystinuria, performed under stringent anticoagulant therapy and provided all special precautions to prevent hypotension, dehydration and stasis of blood are adhered to strictly [18,54-55].
Central nervous system involvement
Mental retardation is the most frequent central nervous system abnormality and often is the first recognizable sign of CbS deficiency [9]. It may present as developmental delay but rarely manifest before the first to second year of life. When walking does begin, a waddling or “Charlie Chaplin-like” gait is usually present [30] and often these children are referred to the orthopaedic surgeons for consultation. Frank mental retardation, when present becomes obvious in middle childhood and beyond [39]. There is a wide variation in IQ reported (median 64; range 10-138) and in general, pyridoxine responsive patients are less susceptible to mental capacity impairment than non-responding counterparts [20]. Twenty-two% of pyridoxine responders had IQ of >90 compared to only 4% of pyridoxine nonresponders. When compared to their unaffected siblings, 34% of pyridoxine responders had comparable IQs with their siblings and a further 63% had lower IQs. Only 6% of pyridoxine nonresponders had comparable IQs to their unaffected siblings with the remaning 94% having lower IQs [20].
About 21% of CbS deficient patients not treated from early infancy have had seizures, grand mal type accounting for 70% of it [9]. Electroencephalographic abnormalities have been reported in cases of CbS deficiency, both with and without a history of convulsion. Excessive slow waves activity [25,49], is the most frequent finding, along with spikes or sharp waves discharges [9,21,56].
Abbott et al looked into the psychiatric manifestations among CbS deficient individuals and found that four categories of mental illness predominates: episodic depression (10%), chronic disorders of behaviour (17%), chronic obsessive-compulsive disorder (5%), and personality disorder (19%). Contrary to earlier anecdotal reports suggesting that schizophrenia may be common amongst this group [14,43,51], Abbott et al did not find sufficient evidence to establish a diagnosis of schizophrenia in any of the 63 individuals studied [57]. Clinically significant psychiatric illness was found in 51%.
Differential diagnosis
The differential diagnosis includes all causes of ectopia lentis, developmental delay/mental retardation, osteoporosis and vascular events, both arterial and venous, particularly when it is found in young people. Deproteinised amino acid profiles and total homocysteine must be included as part of a systematic work-up for these patients, even if the full clinical phenotype is not apparent.
Management and treatment
The aims of treatment in classical homocystinuria vary according to the age of diagnosis. If CbS deficiency is diagnosed in the newborn infant, as ideally it should be, the aim then must be to prevent the development of ocular, skeletal, intravascular thromboembolic complications and to ensure the development of normal intelligence. On the other hand, if the diagnosis is made late when some recognized complications have already occurred, then the clinician’s goal must be to prevent life-endangering thromboembolic events and to prevent further escalation of the complications already suffered [1]. To achieve these clinical aims in treatment, one must try to achieve control or elimination of the biochemical abnormalities characterized by CbS deficiency.
There are currently three recognized modalities of treatment [58]:
1. Pyridoxine,
2. Methionine restricted, cystine supplemented diet, and
3. Betaine, a methyl donor.
Each of these modalities of treatment will be discussed here briefly.
Pyridoxine trial to ascertain responsive status [59]
In-vivo pyridoxine responsiveness does not always correlate to in-vitro responsiveness. Hence, all newly diagnosed patients must be given a pyridoxine trial while remaining on a normal diet. Pyridoxine 50mg three time a day (tds) in the neonate and 100-200mg tds in older children is prescribed to assess vitamin responsiveness with deproteinised amino acids monitoring every three days initially until stabilised. Biochemically, vitamin responsiveness is indicated by a falling homocysteine and methionine levels while remaining on pyridoxine, in the presence of adequate vitamin B12 and folate. While on pyridoxine treatment, the patient is deemed vitamin responsive when the free homocystine is < 5 mmol/L [60]. However, if the free homocystine and methionine remains persistently elevated or rises while on pyridoxine, the patient is biochemically pyridoxine non-responsive and is commenced on a methionine restricted diet.
The status of vitamin responsiveness in a neonate may be difficult to determine on biochemistry alone. While on a trial of pyridoxine, a falling homocysteine and methionine levels due to coincidental growth spurt, may be mistakenly attributed to vitamin responsiveness.
Pyridoxine
Approximately 50% of CbS deficient patients respond to pharmacological doses of pyridoxine and this responsiveness correlates to the presence of residual CbS activity in the cultured fibroblast. Pyridoxine responsiveness is not due to the overcoming of a depletion or defective metabolism of B6 in these patients. The pyridoxine dose required to achieve successful biochemical control is titrated against plasma homocysteine levels. The optimal dose is reached when at a minimum dose of pyridoxine, the plasma homocysteine is at it’s lowest or undetectable.
Doses of pyridoxine required for a response varies markedly among pyridoxine responders. Barber and Spaeth achieved responses to doses of 250 to 500 mg/day [61] while Gaull utilized 800-1200 mg/day of pyridoxine for similar response [62]. However, Perry et al, pointed out that “a patient should not be considered unresponsive to pyridoxine until a dose of 500-1000 mg/day has been given for a period of several weeks” [1]. In general, for older children, a dose of at least 150 mg/day of pyridoxine has been used, although an occasional patient has responded to only 25 mg/day [14]. More recently, Wilcken et al has reported on doses of not more than 200mg/day achieving excellent control together with the appropriate additional therapy [63]. Yap and Naughten in Ireland have used pyridoxine 50 mg tds in the neonate and for the older children, a dose of 100-200 mg tds [60].
Patient safety in the usage of these mega doses of pyridoxine has always featured in the minds of clinicians managing these cases. To date there are no reported side effects from the usage of high dose pyridoxine (up to 500mg/day) in classical homocystinuria, however, 7 non-homocystinuria adult cases have been reported with ataxia and sensory neuropathy on the misuse of megadoses of pyridoxine of 2 to 6 grams daily for 2 to 40 months [64]. Such symptoms improved on withdrawal of pyridoxine.
It is appropriate here to remember that a biochemical response to pyridoxine may not manifest in a potentially responsive patient if folate depletion is present [65] and that patients not given folate while on pyridoxine becomes folate-depleted unless supplemented, but the optimal dose has not been established [64,66]. Centers in Sydney, Nijmegen, Dublin, Manchester and London, however, have used folate 5 mg daily [67].
For those that are responsive to pyridoxine, it remains debatable as to whether pyridoxine alone or in combination with other measures such as methionine restricted, cystine supplemented diet or betaine, a methyl donor, provides the optimal biochemical control. As shown by Boers et al, even the patients with maximal responsiveness to pyridoxine have reduced tolerance to methionine during methionine loading tests and pyridoxine only corrects the biochemical abnormalities in the fasting state [40]. Such patients, in theory, may experience abnormal episodic surge in methionine or homocysteine following protein ingestion [9]. Hence, it follows that these patients may benefit from some methionine restriction or the use of small frequent feedings [9,68] and Wilcken et al showed that the addition of betaine blunted the plasma total homocyst(e)ine in response to these methionine loads [69]. They further suggested that betaine should be added to the treatment regimen of pyridoxine and folic acid in pyridoxine-responsive patients so that the homocysteine accumulation will be normal throughout the day during normal dietary intake [9,69].
Methionine restricted, cysteine supplemented diet
An early diet consisting of methionine restriction and cystine supplementation was devised by Komrower et al [70] and Perry et al in 1966 [71]. Komrower treated a newborn sibling of two affected children allowing a daily methionine intake of 24 to 42 mg/kg body weight/day during the first year of life and about 15 to 25 mg/kg/day during the second year. The plasma methionine was titrated to normal or near normal values during treatment. The daily cystine intake was increased from 25 mg/kg to 73 mg/kg and then to 133 mg/kg of body weight, but the plasma cystine remained considerable less than normal [72]. After 2 years, a daily methionine intake of 10-15 mg/kg body weight reduced the plasma methionine and homocystine to normal or near normal. Despite poor dietary control at home, this child was judged to be clinically normal or near normal at 6.5 years old when last described in 1970 [73].
Perry et al devised a similar diet initially as treatment for a newborn infant, again a sibling to two known affected children [71]. The diet entails reducing daily methionine intake to approximately 20-25 mg/kg body weight for infants, to 10-15 mg/kg body weight for growing children, and to 8-10 mg/kg for adults. L-cystine supplementation of 100-300 mg/kg body weight per day was instituted [1]. They found that with this diet, the patient’s plasma methionine showed dramatic reduction with continued presence of homocystine and continuously subnormal plasma cystine despite supplementation. However, the plasma cystine did approach normal on the occasions when the plasma homocystine has been unusually low [1]. This patient at 7.5 years old had normal growth and none of the recognized complications associated with the condition, a marked contrast to his two other affected siblings in whom had developed severe clinical disease by the age of 6 months [1]. These children were not pyridoxine responsive.
Amelioration of biochemical findings was also noted in those late detected cases in whom some clinical disease may be already present when a methionine restricted cystine supplemented diet was commenced [1,72-6]. As to be expected there was no reversal of the major abnormalities already present before the commencement of dietary treatment, however, further progression of complications seems to be halted or ameliorated.
The parameters recommended to be monitored during dietary treatment are normal growth rate, methioninaemia (< 40 mmol/L) and normal plasma homocyst(e)ine. Plasma cystine should be maintained within the normal range of 67 ± 20 mmol/L and supplemented accordingly (up to 200 mg/kg/day) [29]. Currently, proprietary formulas based on a methionine-free synthetic mixture supplemented with cystine are virtually in exclusive use [9,60].
Betaine-A methyl donor
In pyridoxine-nonresponsive patients, especially when treatment is started late, it is difficult to obtain good compliance to the diet [29,75,78-80]. Perry et al in 1968 first noted in a pyridoxine nonresponsive patient that the administration of choline, a precursor of the methyl donor betaine, decreased homocystine and increased methionine concentrations [76]. Komrower et al has found similar effects using betaine [72]. Other workers have similar effects in response to betaine in pyridoxine nonresponsive patients, namely a reduction in plasma homocyst(e)ine with a rise in plasma methionine in most cases [80-82] and plasma cyst(e)ine rises [72,81] usually in parallel with the reduction in homocyst(e)ine [83].
Betaine is presumed to produce its biochemical effects by increasing the rate of homocysteine remethylation by betaine-homocysteine methyltransferase as more substrate for this reaction is made available. The resultant hypermethioninaemia does not appear to influence the pathophysiology of the disease and has so far not been reported to have any side effects in the literature. More recently, Wilcken et al reported that betaine used as an additional therapy is safe and effective for at least 16 years [63]. Hence, betaine may be useful in pyridoxine nonresponsive patients who will not tolerate methionine restriction or as an adjunct to such a diet [9]. Betaine is usually prescribed at 4 – 6 g/day divided into three daily doses [80,82].
Additional measures
Since foods of animal origin, the major dietary sources of vitamin B12, are excluded from a low methionine diet, adequate B12 must also be provided [1]. Other vitamin and mineral supplement should also complement the diet.
Harker et al reported that pyridoxine in pyridoxine responsive cases and dipyridamole, a platelet aggregation inhibitor, in pyridoxine nonresponsive cases normalized decreased platelet survival and showed a marked decrease in vascular intimal lesions [84-8]. It is from these data that they recommended treatment with dipyridamole 400 mg daily or with the combination of dipyridamole 100mg daily and aspirin 1g daily. While on this regimen, they showed that two patients were free from thromboembolic events in the three years following the commencement of this regimen, whereas there were at least 5 such events in these patients in the year prior to the start of therapy. However, Schulman et al reported on 2 pyridoxine nonresponsive patients who suffered thromboembolic events while on this regimen [88,89].
As thromboembolic events are the major cause of morbidity and mortality in CbS deficient patients, it is perhaps wise to avoid activities associated with an increased risk of thromboembolism, e.g. the use of oral contraceptives and perhaps, even pregnancy. Several workers have reported on the deleterious vascular effects shortly after the start with oral contraceptives in females with CbS deficiency [28,91-3]. More recently, in normal women taking a monophasic sub-50 oral contraceptive there has been a small but significant rise in fasting total homocyst(e)ine during the low-hormone phase of the treatment cycle [9,93].
Diagnosis and diagnostic methods
There are now several tests available for the diagnosis of classical homocystinuria with different sensitivity and specificity.
Urine
The most consistent biochemical abnormality is homocystinuria, as to date there has been no untreated CbS deficient cases described who did not have homocystinuria beyond the period of early infancy [9]. The cyanide-nitroprusside test, the so-called “Brand test”, is a qualitative screening test for the presence of homocystine in the urine, though not totally sensitive or specific. It entails adding 12 drops of a 5% aqueous solution of sodium cyanide to a small test tube containing about 1ml of urine. The tube is agitated to mix then allowed to stand for 5 minutes, followed by the addition of one drop of 5% aqueous solution of sodium nitroprusside. The immediate change of colour from pink to beet-root colour indicates the excessive presence of a disulphide compound in the urine [94-95]. This positive result also occurs in acetonuria, in cases of high urinary creatinine concentration [21], in patients taking penicillamine, those with cystinuria, and in bacterial contamination of urine in cystathioninuria [96-97]. Further tests using high voltage electrophoresis, thin paper or layer chromatography [98] must be undertaken for positive identification of homocystine [30]. This classical test has low sensitivity and has been reported to give false negative results of up to 30% of homocystinuria patients [99]. A modification to this test described by Spaeth and Barber as the “silver-nitroprusside” test, which excludes the detection of cystinuria, may enhance the specificity, but not the sensitivity as these authors claimed [100].
Urinary amino acid analysis using column chromatography is less indicative than in blood. Hypermethioninuria is not completely obligatory in CbS deficiency and homocystinuria on its own does not confirm CbS deficiency, however, if both are concomitantly present then it is conclusive. Quantitative urinary amino acid analysis in CbS deficiency may also reveal the presence of abnormal amounts of other sulphur-containing compounds, including the mixed disulphide of cysteine-homocysteine, homolanthionine [1], S-adenosylhomocysteine[101], and 5-amino-4-imidazole-carboxyamide-5’-S-homocysteinylribonucloeside [102].
Blood
It is essential for the diagnosis of classical homocystinuria to be confirmed by the determination of amino acids in fasting blood [1]. The biochemical hallmark of this condition is the presence of homocystine in the plasma in association with increased levels of homocysteine-cysteine mixed disulphide and in combination, almost consistently (except for 6% of cases), with hypermethioninaemia and hypocystinaemia.
Accurate technique is crucial. In particular, the blood sample need to be deproteinised promptly, within 10 minutes, if underestimation of serum free Hcy is to be avoided due to the binding of sulphydryl and disulphide amino acid by disulphide interchange to the sulphydryl groups of plasma proteins. It is also important that the customary norleucine not be added to the sample as an internal standard in plasma amino acid analysis of homocystinuria as the mixed disulphide of cysteine and homocysteine is eluted at the same point making accurate quantitation impossible. g-aminobutyric acid or homoarginine, as well as other unusual amino acids, can be used as an internal standard. Rapid amino acid analyser systems that fails to separate homocystine from the ammonia peaks should also not be used in establishing a definitive diagnosis of classical homocystinuria or in biochemical response to treatment [103]. If total Hcy is the measurement used in diagnosis, it is again imperative to separate the blood cells from the blood sample rather quickly to avoid inaccurate results.
Direct enzyme assays
Further and definitive confirmation of a diagnosis of CbS deficiency is by assaying the CbS enzyme activity in liver biopsy, cultured skin fibroblast, phytohemagglutinin-stimulated lymphocytes or long-term cultured lymphocytes. The findings of not detectable or up to 15% of mean control level of CbS enzyme activity measured in cultured skin fibroblast provides the final diagnosis [104-110], and will differentiate the 6% of the CbS deficiency cases with hyperhomocysteinaemia and normal methionine from cases of remethylation difficulties.
Molecular diagnosis- CbS Mutational analysis
In addition to the above tests, CbS mutational screening could be another option, with the technology being more and more available as the library of known CbS mutations enlarges. There are now more than 92 known disease causing CbS mutations [108] the two most common mutation, the G307S, also known as the “Celtic” mutation, heralds the more severe pyridoxine non responsive phenotype. The other common mutation is the I278T which is associated with the milder pyridoxine responsive type.
Antenatal diagnosis
Prenatal diagnosis of CbS deficiency has been feasible since Fowler et al reported the first diagnosis in an affected fetus in 1982 [112]. Extracts of cells from cultured amniotic fluid contains readily detectable activity of CbS [104,108,113-116]. Not only the homozygous state can be detected prenatally, several workers have reliably diagnosed the obligate heterozygote states of fetuses from homozygous mothers [110,115,117]
The catalytic activity of CbS in chorionic villi is insufficient for direct assay of the enzyme, however, using extracts of cells grown in tissue culture from chorionic villi, one CbS deficient fetus and two unaffected fetuses have been correctly diagnosed before the 12th week of gestation [9 ]. Hence, classical homocystinuria is one of the inherited metabolic disorders that can be reliably diagnosed in-utero.
Clinical outcome-Effect of chronic treatment
If untreated, the prognosis of an individual diagnosed with classical homocystinuria is indeed very bleak with progressive morbidity and mortality as documented by Mudd et al in 1984 [20]. There is now established evidence that treatment of the condition does prevent complications from developing or ameliorate complications already suffered before treatment, which ever may be the case. Treatment, however, does not reverse complications already suffered. Recently, Wilcken et al reported in a late-detected group that “ treatment which effectively lowers circulating homocyst(e)ine, even to suboptimal levels, markedly reduces cardiovascular risk in patients with CbS deficiency” [63].
The efficacy of dietary treatment in the early treated patients on preventing ectopia lentis, mental retardation and especially radiographical evidence of osteoporosis and thromboembolic events was reported by Yap and Naughten [60]. The Irish center reported on a lifetime free homocystine median of <11 mmol/L in those early treated, newborn screened pyridoxine non-responsive patients (n=15) with no complications. They all had vision of 20/20 [118]. The full scale IQ ranged from 84 –120 (mean 105.8) in 13 of the Irish early treated and compliant patients (mean age 14.4 yrs; range 4.4-24.9). These patients also had comparable IQs to their unaffected siblings (n=10, mean age 19.4; range 9.7-32.9) with mean full scale IQ of 102 (range 76-116) [119]. Similar findings were also reported in 11 pyridoxine non-responsive early treated patients in Manchester, England with median IQ of 100 (range 84-117), significantly better than the median of 58 (range 20-86) amongst those late diagnosed [120]. There were no documented thromboembolic events amongst the Irish patients with 403.9 patient-years of treatment [121]. Homocysteine-lowering treatment was also shown to reduce the vascular risk significantly, despite imperfect biochemical control, in a multicenter study involving 158 patients with 2822 patient-years of treatment 67.
Classical homocystinuria is a potentially treatable condition, especially if detected and treated early. Total homocysteine should be included in the work-up of patients with ectopia lentis, mental retardation, premature vascular events and osteoporosis.
Homocystinuria is inherited as an autosomal recessive trait, which means that the child must inherit the defective gene from both parents to be seriously affected. Usual findings in homocystinuria are nearsightedness, dislocation of the lens of the eye, and a tendency to develop blood clots in the veins and arteries.
Newborn infants appear normal, and early symptoms, if present at all, are vague and may occur as mildly delayed development or failure to thrive. Increasing visual problems may lead to diagnosis of this condition when the child, on examination, is discovered to have dislocated lenses and nearsightedness.
Some degree of mental retardation is usually seen, but some affected people have normal IQs. When mental retardation is present, it is generally progressive if left untreated. The condition can also increase the risk for psychiatric disorders.
Homocystinuria has several features in common with Marfan syndrome, including dislocation of the lens; a tall, thin build with long limbs; spidery fingers (arachnodactyly); and a pectus deformity of the chest. In addition, affected people may have high arches of the feet (pes cavus), knock-knees (genu valgum), and a curved spine (scoliosis).
Affected people commonly develop blood clots. These clots can dislodge and travel (i.e., form an embolus) and damage any tissue in which the clot lodges. Clots that travel to the brain can cause stroke, for example.